单细胞 ATAC-seq(scATAC-seq)能够捕获单细胞水平的染色质开放信息,是用于研究基因调控和细胞异质性的重要方法之一。细胞注释是 scATAC-seq 数据分析中非常重要的一步,然而,scATAC-seq 数据由于其高维度、高稀疏度、高噪音的特点,使得细胞注释较为困难。大多数现有的注释方法单纯基于基因活性,这有可能会忽略大量基因间区的信息。还有一些方法基于多模态整合后的标签转移,这容易受到批次效应的影响,并且可能会忽视稀有的细胞类型。
2023年7月26日,国际知名生物信息学学术期刊Briefings in Bioinformatics在线发表了mg4355娱乐线路检测官网田卫东教授课题组题为“AtacAnnoR: a reference-based annotation tool for single cell ATAC-seq data”的研究论文。作者开发的一种新颖的单细胞ATAC-seq数据的细胞注释算法AtacAnnoR。AtacAnnoR可以利用已标注的 scRNA-seq 数据作为参考,能够对 scATAC-seq 的细胞类型进行精准的细胞注释。
AtacAnnoR利用两轮注释的方法,从而避免批次效应并实现跨模态细胞注释。第一轮注释主要是在基因层面的注释,基于基因活性与基因表达的相似性,结合严格的筛选策略,筛选出部分高可信的种子细胞。第二轮注释基于整个基因组组染色质开放的相似性,利用种子细胞注释剩余细胞。AtacAnnoR的创新点在于:由于种子细胞本身就来源于待注释的细胞,因此不受批次效应的影响,其次,方法利用了整个基因组peak的信息,相对于只利用基因层面的信息会更加准确。
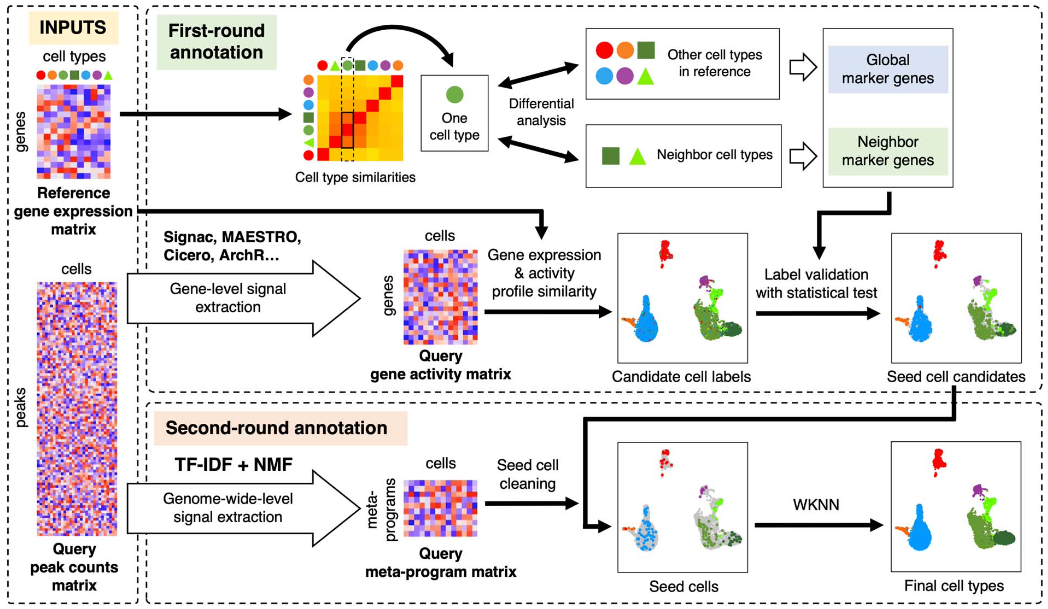
算法流程
作者设计了三种情况,系统地对 AtacAnnoR 的表现进行了测试。这三种情况分别是:
细胞层面的双组学测序数据(cell-level dual omics sequencing)。即在同一个细胞内同时测量基因表达和染色质开放,这种情况可以作为金标准来验证 scATAC-seq 细胞注释工具的准确性。
样本层面的双组学测序数据(sample-level dual omics sequencing)。即同一份样本分成两份分别进行 scRNA-seq 和 scATAC-seq。这种数据通常是研究人员为了自己的研究目的从而进行了特殊的实验设计,对双组学分别进行测序。
仅有待注释的 scATAC-seq 数据,使用其他来自公共数据库的 scRNA-seq 作为参考来进行细胞注释。这种情况是最普遍,同时也是难度最大的一种情况。因为大多数情况下并没有配套的 scRNA-seq 作为参考,公共数据库的 scRNA-seq 数据可能会与手上的 scATAC-seq 数据存在较大的批次效应。
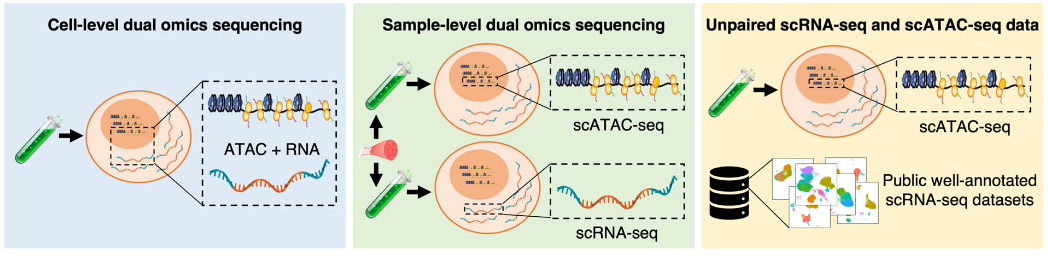
测试数据集的三种情况
作者将AtacAnnoR 和Seurat v3(2019, Cell),GLUE(2022, Nature biotechnology),scJoint(2022, Nature biotechnology),Conos(2019, Nature methods), MAESTRO(2020, Genome biology)和 CellWalkR(2021, Genome biology)进行了比较。
在前两种情况下,AtacAnnoR 的注释准确率和 GLUE 几乎处于并列第一的位置,而平衡准确率(balanced accuracy)要远好于其他方法,说明 AtacAnnoR 不止能对数量多的细胞类型准确注释,同时也能关注到细胞数量较少的亚群。作者对稀有细胞类型的准确率检查也说明可这一点:AtacAnnoR在总共32种稀有细胞类型中的29种上的注释准确率都为第一,领先第二名方法将近20个百分点。Seurat v3 和 scJoint 是表现也还不错的方法,但 Seurat 在细胞比例极端不平衡的数据集上表现不佳,而 scJoint 的问题在于对稀有细胞类型的注释效果不佳。
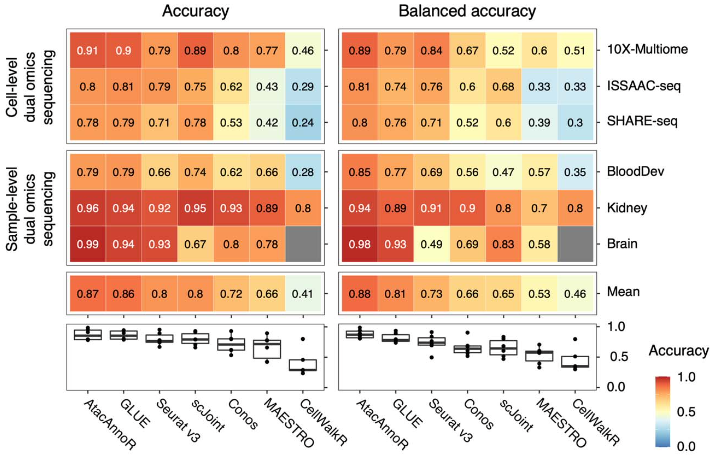
前两种情况的AtacAnnoR与其他方法注释结果比较
对于第三种情况,AtacAnnoR 的优势更加明显,达到了 0.91 的平均准确率,而第二名的 Seurat v3 仅有 0.75。在前两种情况表现很好的 GLUE 方法在地三种情况下仅达到了 0.55 的准确率。这说明其他方法受批次效应的影响较大,而AtacAnnoR 几乎不受影响。
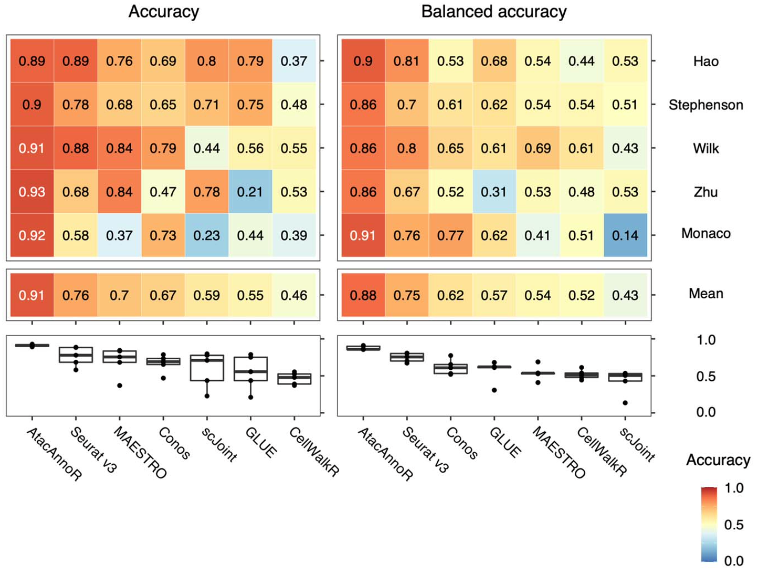
第三种情况的AtacAnnoR与其他方法注释结果比较
作者调查了其他方法失败的可能原因。作者发现,GLUE 注释出的 scATAC-seq 的细胞比例与 scRNA-seq 参考数据集的细胞比例有着非常高的相关性,Seurat v3 也有部分相关性,这可能是因为他们都是首先对两个模态进行数据整合,然后再利用近邻细胞进行细胞注释。当参考数据和待注释数据的细胞比例有较大差异时,整合可能失败,从而导致细胞注释结果不准确。
此外,作者还考虑到实际应用中有可能面对多套参考数据的情况。当多套参考可用时,作者开发了一种综合多套注释结果的策略,可以继续提高注释准确率3-5个百分点。这进一步增强了算法在实际应用中的价值。
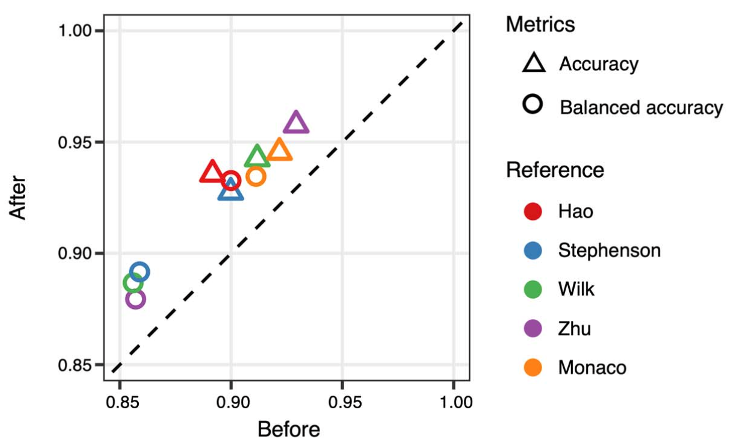
综合多套参考数据的注释结果进一步提高注释准确率
最后,AtacAnnoR已经被包装成一个简单易用的R语言软件包,方便用户使用。软件具有两种模式:默认模式可以使用一行代码完成细胞注释,并且可在当前常用的scATAC-seq分析流程中直接调用;精调模式可以让用户使用内置的20多种函数一步步可视化中间结果,并对参数进行调整。最后,AtacAnnoR的速度与其他方法相比也是最快的,能够在5分钟内注释超过10万个细胞。
工具链接:https://github.com/TianLab-Bioinfo/AtacAnnoR
综上,作者开发了基于两轮注释的scATAC-seq的细胞注释算法AtacAnnoR;与竞争方法相比,AtacAnnoR优势显著,尤其是对稀有细胞类型的注释和批次效应较大的情况下。该工作提供的软件包能够帮助研究者更加准确快速地注释scATAC-seq细胞类型,有助于帮助scATAC-seq下游分析提供更有意义的生物学发现。
mg4355娱乐线路检测官网计算生物学系博士生田乐进第一作者,mg4355电子娱乐官网田卫东教授为通讯作者。该工作得到了国家自然科学基金和国家重点研发计划的支持。
原文链接:https://doi.org/10.1093/bib/bbad268