线粒体DNA耗竭综合征(Mitochondrial DNA Depletion Syndrome,简称MTDPS)是一组罕见的遗传疾病,其特点是患者细胞内的线粒体DNA含量显著降低。MTDPS可以分为多个亚型,其中最常见的亚型是由于缺乏线粒体DNA复制酶或DNA修复酶所导致的。这些酶的缺陷会干扰线粒体DNA的复制和修复过程,使得线粒体内的DNA逐渐减少。由于线粒体DNA在编码线粒体蛋白质所需的基因中起着关键作用,MTDPS患者往往出现线粒体蛋白质的缺乏或功能异常。这会导致细胞能量代谢受损,进而影响多个器官和组织的功能。临床表现方面,MTDPS患者常常表现为全身性疾病,包括肌无力、肝功能异常、神经系统障碍等。病情的严重程度和症状的种类可以因亚型的不同而有所差异。
Cullin家族E3泛素连接酶底物亚基—FBXL4是新发现的一个MTDPS亚型(MTDPS13)致病基因。病人的临床表型主要表现为高乳酸血症,发育迟缓和肌张力减退为特征的多系统疾病,其他特征包括喂养困难、小头畸形、高氨血症、癫痫发作、肥厚型心肌病、肝转氨酶升高、复发性感染和面部特征多样等。目前尚无有效的治疗策略。因此,阐明FBXL4突变致病的分子机制,寻找有效的靶向干预治疗手段是亟待解决的临床难点问题。
2023年8月12日,mg4355娱乐线路检测官网王陈继团队、基础医学院马丽香团队、附属妇产科医院赵世民团队和上海市儿童医院陈育才团队合作在Cell Death & Differentiation在线发表了题为 FBXL4 mutations cause excessive mitophagy via BNIP3/BNIP3L accumulation leading to mitochondrial DNA depletion syndrome的研究论文(图一)。该研究发现FBXL4基因突变导致线粒体自噬(mitophagy)受体BNIP3和BNIP3L蛋白异常积累,线粒体自噬过度激活,进而导致线粒体数目急剧下降,是其突变致病的内在分子机制。

鉴于FBXL4是一个Cullin家族E3泛素连接酶底物亚基,研究人员首先猜测FBXL4可能参与了和线粒体稳态维持相关蛋白的泛素化修饰调控。通过蛋白质亲和纯化质谱技术,研究人员发现BNIP3和BNIP3L是FBXL4的潜在互作蛋白。BNIP3和BNIP3L属于BCL-2家族的成员,是定位在线粒体外膜的膜蛋白。它们的转录表达受到低氧诱导因子HIF1α所调控。当细胞处于低氧状态时,BNIP3和BNIP3L的表达水平上调。它们通过与自噬核心蛋白LC3/GABARAPs结合,促进线粒体与自噬体的结合并降解。这种选择性的线粒体自噬有助于维持细胞内线粒体的数量和和功能,从而维持细胞的稳态。
后续的实验证实,BNIP3/3L蛋白的半衰期很短,泛素化降解作用活跃。FBXL4可以和BNIP3/3L特异性结合,并促进其通过蛋白酶体途径的泛素化降解。细胞中FBXL4敲除导致的线粒体数目显著下降和代谢异常表型可以被同时敲减BNIP3/3L的表达所逆转。MTDPS13病人来源的FBXL4错义突变体不能正确组装成CUL1-RBX1-FBXL4 E3泛素连接酶复合体,导致BNIP3/3L的泛素化降解受阻。异常积累的BNIP3/3L导致细胞在非应激条件下即存在过度的线粒体自噬,线粒体数目显著下降,从而导致患者神经系统在内的多系统异常临床表型(图二)。
为了进一步确证上述分子机制,研究人员从一名FBXL4移码突变(p.L332Tfs*3)的MTDPS13患者获取外周血单核细胞,然后通过重编程技术诱导成多能干细胞(iPSCs),然后将iPSCs分化为神经前体细胞(NPCs)或成熟皮质神经元(Cortical Neurons)。进一步检测发现, FBXL4突变的神经前体细胞或皮质神经元中,BNIP3/3L蛋白异常累积,并且表现出的远高于对照的线粒体自噬发生。此外根据病人突变位点构建的位点敲入小鼠模型也证实FBXL4突变导致小鼠脑、心、肝和肺等多器官中BNIP3/3L蛋白异常累积和线粒体数量显著降低。在FBXL4突变小鼠的成纤维细胞(MEFs)中同时敲低BNIP3/3L的表达即可逆转自发性的高水平线粒体自噬。由此,在小鼠模型和病人来源的iPSCs诱导分化的神经细胞体系得到的结果进一步支持了上述分子机制。
最后,研究人员讨论了该病潜在的靶向治疗手段。鉴于BNIP3/3L的异常积累是MTDPS13的致病原因,后续可通过PROTAC技术设计并合成促进BNIP3/3L的靶向降解的小分子,降低突变细胞内的BNIP3/3L的蛋白水平来抑制过高的线粒体自噬,从而实现靶向治疗的目的。此外,某些已知的抑制线粒体自噬的小分子,如mDivi-1等,也有拮抗BNIP3/3L积累导致的高水平线粒体自噬的应用潜力。
总之,这项工作将从“FBXL4-BNIP3/3L-mitophagy”调节轴的全新角度诠释FBXL4突变引起线粒体DNA耗竭综合征的分子机制,为后续的靶向治疗提供了坚实的理论依据。
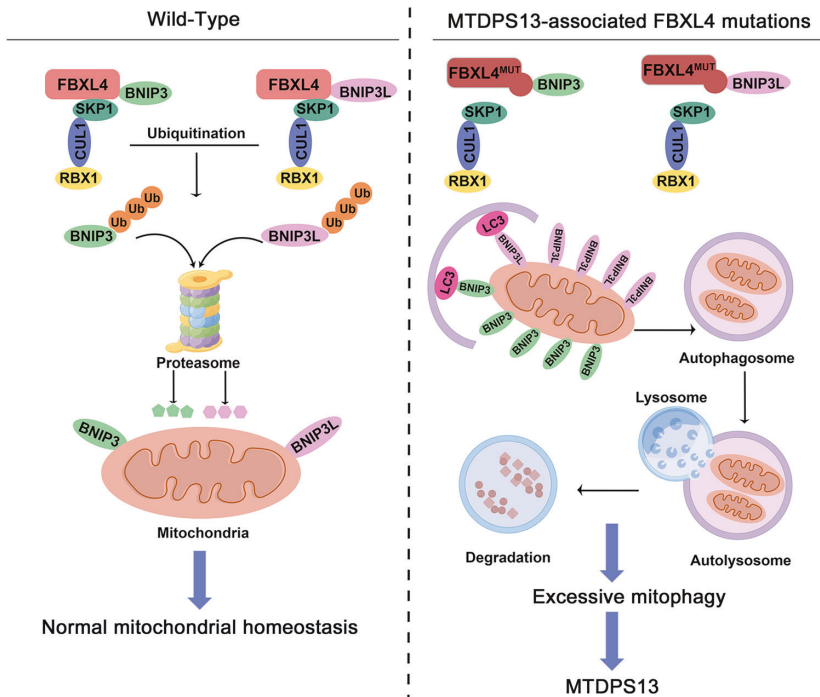
图二、FBXL4突变导致线粒体DNA耗竭综合征13型的分子机制。
mg4355娱乐线路检测官网博士生陈莹佶、焦冬月,mg4355电子娱乐官网基础医学院硕士生刘阳为该论文第一作者。王陈继、马丽香、赵世民和陈育才为该文的通讯作者。
原文连接:https://www.nature.com/articles/s41418-023-01205-1